Our mission is to improve human health through the development of precision medicine technologies to study and diagnose infectious, immune and microbiome-associated disease. Our research brings approaches from engineering, biophysics, and computational biology to genomics and medicine.
We pursue research in two areas:
A) Liquid biopsies for infectious and immune-mediated disease: We investigate technologies and applications of circulating cell-free DNA in diagnostic medicine. We have developed molecular assays to obtain rich, sequence information from cell-free DNA in blood and urine, and computational methods to classify infectious and immune-mediated disease from cell-free DNA ‘omics data. We have demonstrated applications of these technologies in the monitoring of infection, host tissue injury due to infection, COVID-19, solid-organ transplant rejection, and graft-versus-host disease.
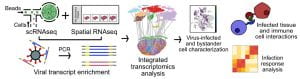
B) Digital spatial profiling of microbiomes and infection in complex tissues: We investigate single-cell and spatial sequencing technologies to study infection in native, complex tissues and to spatially profile bacterial microbiomes and host-microbiome interactions.
Liquid biopsies for infectious and immune-mediated disease
Overview. Short fragments of cell-free DNA and RNA circulate in blood, urine and other biofluids and offer an information-rich window into human physiology. We pursue technologies and applications of cell-free nucleic acids as liquid biopsies for infectious and immune-mediated disease. 
Urinary cfDNA is a versatile analyte for the diagnosis of urinary tract infection. In recent work, we have explored the utility of urinary cell-free DNA as an analyte for urinary tract infection, one of the most common infectious complications in humans. We found that genomics-based measurements of the structure and diversity of urinary cell-free DNA can inform the UTI, the urinary microbiome and virome, antibiotic drug-resistance and susceptibility, and bacterial growth dynamics. To assess the clinical utility of this “one stop shop” diagnostic test, we assayed more than one hundred urine samples collected from a cohort of kidney transplant patients. We are evaluating the performance of urinary cell-free DNA as an analyte to diagnose tuberculosis in collaboration with Global Health labs.
A liquid biopsy for infectious disease. High throughput metagenomic sequencing offers an unbiased approach to identify pathogens in clinical samples. However, conventional metagenomic sequencing does not integrate information about the host, which is critical to distinguish colonization from infectious disease. To overcome this challenge, we have developed a novel assay to simultaneously quantify the abundance of a large range of viral and bacterial pathogens, and the degree of host tissue injury due to infection. This is accomplished with a genome-wide measurement of methylation marks comprised within cell-free DNA to trace their tissues-of-origin and quantify the degree of injury to different host tissues.
A liquid biopsy for COVID-19. COVID-19 primarily affects the lungs, but evidence of systemic disease with multi-organ involvement is emerging. We have developed a blood test to broadly quantify cell, tissue, and organ specific injury due to COVID-19, using genome-wide methylation profiling of circulating cell-free DNA in plasma.
Screening of complications of hematopoietic transplantation. More than 30,000 patients undergo allogeneic hematopoietic cell transplants (HCT) worldwide each year for treatment of a variety of malignant and nonmalignant hematologic diseases. Monitoring of post-transplant complications is critical, yet current diagnostic options are limited. We have recently shown that cell-free DNA in blood is a highly versatile analyte for monitoring of the most important complications that occur after HCT: graft-versus-host disease (GVHD), a frequent immune complication of HCT; infection; relapse of underlying disease; and graft failure.
Tool development. We have created a bioinformatics tool to screen for infection from low-biomass isolates of cell-free DNA, approaches to account for length biases in metagenomic cfDNA sequencing assays, a metagenomic DNA sequencing assay that is robust against environmental DNA contamination (unpublished), and a single-stranded DNA library preparation that enables analyses of highly degraded, ultrashort cfDNA in blood and urine. 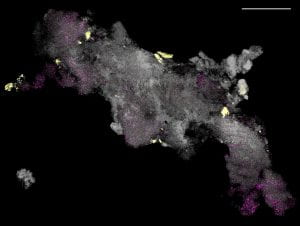
Before Cornell. Iwijn’s research as a postdoctoral scholar in the lab of Steve Quake at Stanford University led to advances in cell-free DNA research that have already impacted human health in a significant way. In transdisciplinary collaboration with a team of heart and lung transplant physicians and bioengineers at Stanford, Iwijn developed a simple blood test for the early and noninvasive diagnosis of rejection after solid-organ transplantation. More than 49,000 patients benefited from this test in 2019 alone. In 2013, Iwijn’s work led to the discovery of the presence of microbial cell-free DNA in human blood. This work suggested that metagenomic sequencing of microbial cell-free DNA could lead to a ‘digital culture’ test that can detect over one thousand bacterial, viral and fungal infections from blood. A commercial version of this assay is already in use in more than 100 US hospitals.
Digital spatial profiling of host-microbiome and host-pathogen interactions
Overview. We investigate single-cell and spatial sequencing technologies to study infection in native, complex tissues and to spatially profile bacterial microbiomes and host-microbiome interactions.
Spatial mapping of microbiomes. Microbial communities exhibit rich species diversity and exquisite spatial organization. While the species diversity of microbial populations is accessible by shotgun DNA sequencing; their spatial organization remains difficult to survey with existing tools. To overcome this limitation, we have developed High Phylogenetic Resolution microbiome mapping by Fluorescence In-Situ Hybridization (HiPR-FISH), a versatile and cost-effective technology that uses binary encoding and spectral imaging and machine learning-based decoding to create micron-scale maps of the locations and identities of hundreds of microbial species in complex communities.
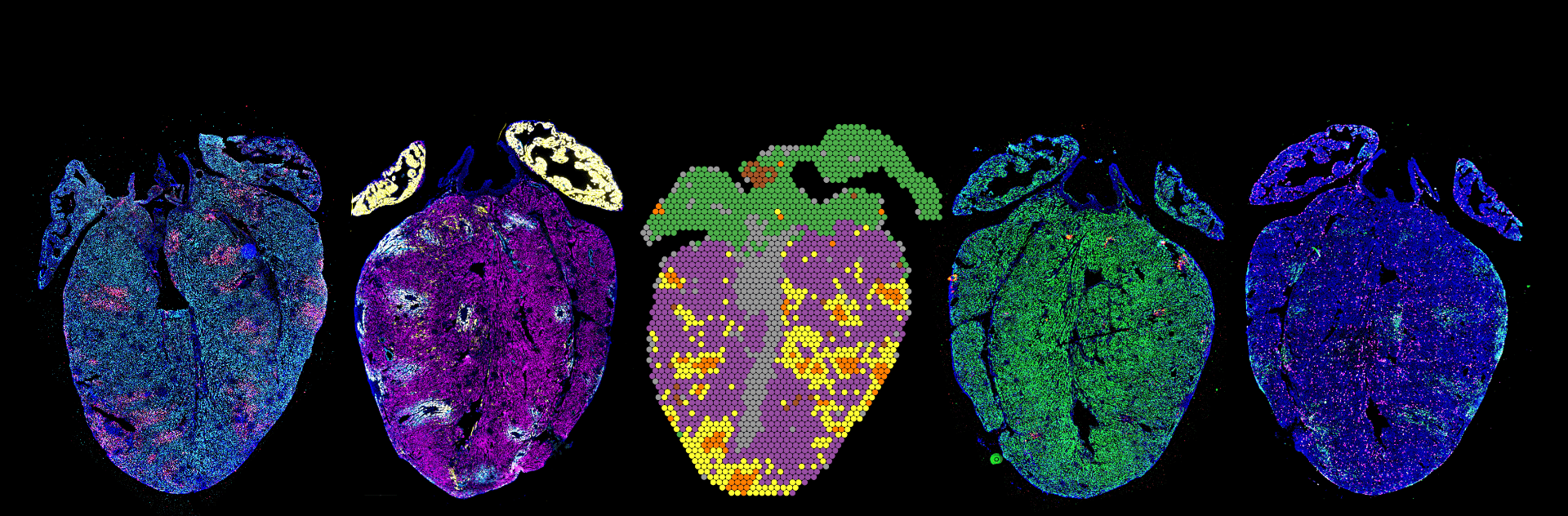
Mapping of infection in complex tissues. Little remains known about how RNA viruses spread systemically and within organs and tissues. We are adopting spatial RNA sequencing technologies, including the 10X Visium platform and custom technologies for spatial RNA-sequencing at a high spatial resolution to map RNA virus infection in complex tissues. We have recently used these approaches to learn about the pathogenesis of myocarditis.
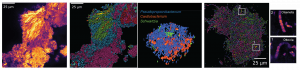
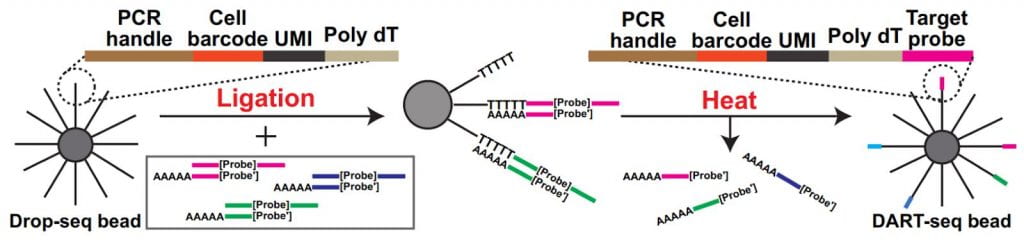
Simultaneous transcriptome profiling and amplicon sequencing in single-cells. High-throughput single-cell RNA sequencing (scRNA-seq) has provided important insights into cellular complexity and transcriptome dynamics. However, current implementations of this technology are limited to capturing information from the ends of A-tailed messenger RNA (mRNA) transcripts. To surmount this limitation, we developed a versatile technology, Droplet Assisted RNA Targeting by Single cell Sequencing, that allows interrogation of all regions of the polyadenylated transcriptome, as well as measurement of other classes of RNA in the cell.
Spatial Total RNA-Sequencing (STRS) Spatial transcriptomics reveals the spatial context of gene expression, but current methods are limited to assaying polyadenylated (A-tailed) RNA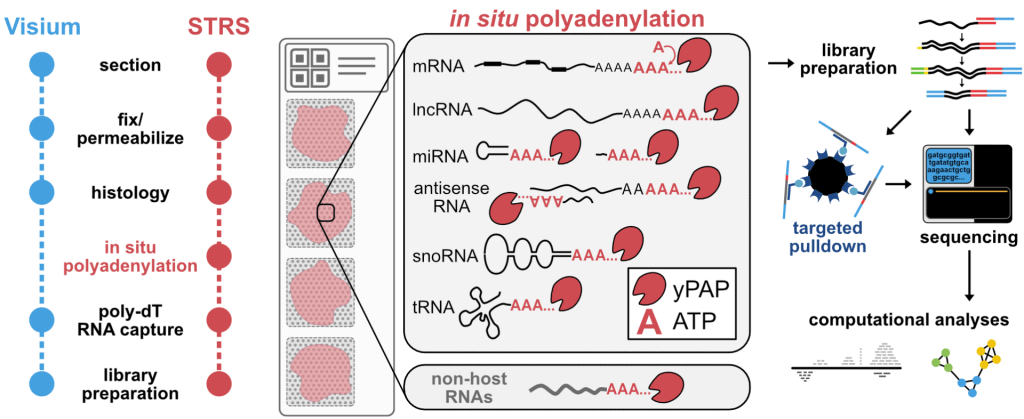 transcripts. We used enzymatic in situ polyadenylation of RNA to detect the full spectrum of RNAs, expanding the scope of sequencing-based spatial transcriptomics to the total transcriptome. In a recent study in Nature Biotechnology, we applied STRS to study skeletal muscle regeneration and viral-induced myocarditis. Our analyses reveal the spatial patterns of noncoding RNA expression with near-cellular resolution, identify spatially defined expression of noncoding transcripts in skeletal muscle regeneration and highlight host transcriptional responses associated with local viral RNA abundance. STRS requires adding only one step to the widely used Visium spatial total RNA-sequencing protocol from 10x Genomics, and thus could be easily adopted to enable new insights into spatial gene regulation and biology.
transcripts. We used enzymatic in situ polyadenylation of RNA to detect the full spectrum of RNAs, expanding the scope of sequencing-based spatial transcriptomics to the total transcriptome. In a recent study in Nature Biotechnology, we applied STRS to study skeletal muscle regeneration and viral-induced myocarditis. Our analyses reveal the spatial patterns of noncoding RNA expression with near-cellular resolution, identify spatially defined expression of noncoding transcripts in skeletal muscle regeneration and highlight host transcriptional responses associated with local viral RNA abundance. STRS requires adding only one step to the widely used Visium spatial total RNA-sequencing protocol from 10x Genomics, and thus could be easily adopted to enable new insights into spatial gene regulation and biology.
Collaborative projects
Because of our expertise in molecular engineering, data science, and single-cell genomics, we often get to participate in collaborative projects.
Muscle development and repair. With Biomedical engineering colleague Dr. Ben Cosgrove we are studying the muscle repair response to injury. As part of this effort, we recently created a large-scale single-cell transcriptomic atlas of mouse skeletal muscle by integration, consensus annotation, and analysis of 23 newly collected scRNAseq datasets and 79 public single-cell (scRNAseq) and single-nucleus (snRNAseq) RNA-sequencing datasets. The resulting datasets includes more than 365,000 cells and spans a wide range of ages, injury, and repair conditions. This effort enabled us to identify and analyze rare, short-lived transitional states of progenitor commitment and fusion that are poorly represented in individual datasets. Our work supports the utility of large-scale integration of single-cell transcriptomic data as a tool for biological discovery.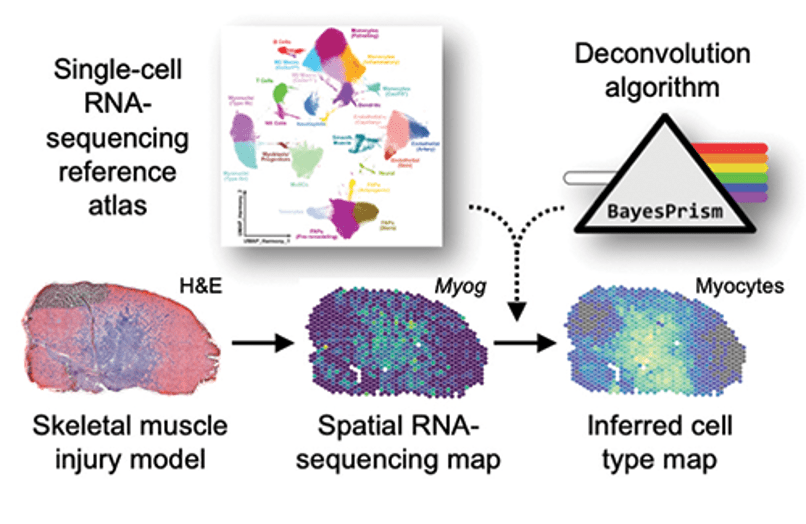
Heart development. With biomedical engineering colleague Dr. Jonathan Butcher we are using spatiotemporal single-cell RNA sequencing to study fetal heart development in a chick model. Together, we discovered TMSB4X persistently enriched in coronary vasculature of chicken embryonic hearts.
Uncovering Transcriptional Dark Matter via Gene Annotation Independent Single-Cell RNA Sequencing Analysis. scRNA-seq expression analyses rely heavily on the availability of a high quality annotation of genes in the genome, and genome annotations often fail to cover the full transcriptome of every cell type through development, in particular for organisms which are not routinely studied, which we revealed with experiments and analyses spanning human, mouse, chicken, naked mole rat, gray mouse lemur and sea urchin. To overcome this hurdle, we created a scRNA-seq analysis workflow that recovers biologically relevant information beyond the scope of the best available gene annotation. This advance led to a collaboration with the Tabula Microcebus consortium at the Chan Zuckerberg Institute and Stanford University. This atlas comprises more than 200,000 single-cell transcriptomes from 27 organs and identifies 100s of mouse lemur cell types.
Reverse tissue-engineering of a vascular graft. With Biomedical engineering colleague Dr. Yadong Wang we are using scRNA-seq as a tool to help to reverse tissue-engineer vascular grafts.